How to Query Ensembl BioMart with Python
Posted on Tue 19 January 2021 in Python
Introduction
Recently, me and my colleagues wrote a manuscript involving meta-analysis of RNA-Seq studies. One of my tasks of this project was to perform a Gene Ontology (GO) enrichment analysis: “[G]iven a set of genes that are up-regulated under certain conditions, an enrichment analysis will find which GO terms are over-represented (or under-represented) using annotations for that gene set”. In other words, I could verify which cellular pathways were in action during the experimental conditions.
After I finished the GO analysis, I got a spreadsheet with a list of GO terms — brief descriptions of the cellular process performed by each pathway. When I was thinking about the pathways, I started to wonder: “which proteins participate into each pathway?” So I decided to go data mining the Ensembl BioMart to find out those protein genes.
Ensembl is a huge project by the European Bioinformatics Institute and the Wellcome Trust Sanger Institute to provide databases of annotated genomes for several (mainly vertebrate) species. BioMart is one of their data mining tools. In this post, I will describe how I used Python to query BioMart. I will introduce a simple function to generate GNU Wget commands to retrieve query results via RESTful access. Then, I will show how I aggregated the data to met my objective (i.e. list all genes participating in a biological pathway represented by a GO term).
The code and example files presented here are available in my portfolio.
Prepare identifiers
I created a project folder where I saved the Python script with this demonstration’s code (ensembl_rest.py) and two subfolders. The data folder holds the example data: a spreadsheet named go_demo.xlsx. The src folder contains the functions’ code. I will write about them later.
.
├── data
│ └── go_demo.xlsx
├── src
│ ├── files_to_pandas.py
│ ├── lists_ensembl.R
│ └── query_biomart.py
└── ensembl_rest.py
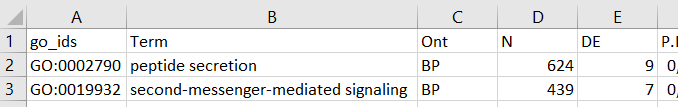
The spreadsheet contains just two rows of data (so that the computation can be completed quickly). They are derived from a GO enrichment analysis output performed by limma‘s package goana function available for R software. The GO ids were the identifiers (search keywords) I used to query BioMart. You may use whatever identifier recognized by BioMart (more on that later). Just be sure they are on a nicely-named column so you can read it into Python.
Load modules and set file paths
These are the modules I used:
import glob
import subprocess
from collections import defaultdict
from pathlib import Path
import dask.dataframe as dd
import pandas as pd
from dask import delayed
from src.files_to_pandas import files_to_pandas
from src.query_biomart import query_biomart
The first four modules are from Python’s standard library. I already used dask before. If you do not have dask or pandas installed, do it now with pip. I installed openpyxl as well, since it serves to read/write Excel spreadsheets.
pip install "dask[complete]" pandas openpyxl
Observe that I am explicitly loading the functions by prefixing the modules names with src. (remember that we can call every Python script a module).
Using pathlib.Path I nicely set the folders and files paths, using the project folder as the root.
project_folder = Path().resolve() # project root
data_folder = project_folder / "data"
go_list = data_folder / "go_demo.xlsx"
Then I loaded the identifiers file into a pandas.DataFrame with the help of openpyxl.
df = pd.read_excel(go_list, engine="openpyxl")
Configure the queries: datasets, filters, values and attributes
Now let’s examine an excerpt from the query_biomart() function code. It contains the necessary modules, the function’s arguments and some of their types hinted with the help of typing module:
import re
from typing import List
def query_biomart(
filter_names: List[str],
values: List[str],
attribute_names: List[str],
dataset_name: str = "hsapiens_gene_ensembl",
formatter: str = "TSV",
header: bool = False,
uniqueRows: bool = False
) -> str:
# ...Function here ...
There is a description of each argument below:
filter_names(List[str]): A list of strings that define restrictions on the query.values(List[str]): A list of strings containing the values that will be used for the filters. Must have same length offilter_names.attribute_names(List[str]): A list of strings that define the data characteristics that must be retrieved for the filtered data.dataset_name(str, optional): A string indicating which dataset will be queried. Each species has their respective dataset. Defaults to “hsapiens_gene_ensembl” (humans).formatter(str, optional): A string to indicate how output must be formatted. Options: “HTML“, “CSV“,”TSV” and “XLS“. Defaults to “TSV“.header(bool, optional): A Boolean indicating if the output must have a header. Defaults toFalse.uniqueRows(bool, optional): A Boolean indicating if the output must have unique rows (deduplicate data). Defaults toFalse.
Thus I searched BioMart:
- Into Homo sapiens dataset (“hsapiens_gene_ensembl”),
- Filtering by GO terms (“go_parent_term”),
- Using GO ids (for example, GO:0002790) as search keywords (values),
- And wanted to retrieve the gene name attribute (“external_gene_name”).
Here is an example of the function with the inputs above. All defaults were maintained, except for uniqueRows, which I set to True to remove duplicate data:
query_biomart(filter_names=["go_parent_term"],
values=["GO:0002790"],
attribute_names=["external_gene_name"],
uniqueRows=True)
See the Appendix to help you see which information can be searched in BioMart.
The output of the function is a string ‐ a ready-to-use GNU Wget Unix program command. The query string inside the command has a XML format that is sent to Ensembl’s servers. The query result will then be saved on a file named based on the values (search keywords) of the query. The extension of the file depends on the formatter argument.
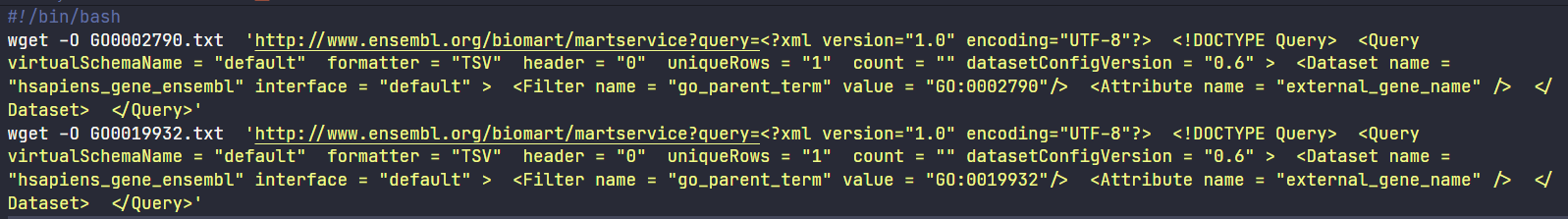
I created a Bash script named commands.sh inside the data folder to backup and document my work. I did this with a for loop. In other words, Python wrote for me a series of Wget commands, one for each keyword in the identifiers data frame (go_ids column ‐ df["go_ids"]) alongside the desired filter and attribute:
with open(data_folder / "commands.sh", "w", newline="\n") as commands:
commands.write("#!/bin/bash" + "\n")
for go_id in df["go_ids"]:
commands.write(query_biomart(filter_names=["go_parent_term"],
values=[go_id], attribute_names=["external_gene_name"],
uniqueRows=True) + "\n")
See a print screen of the commands.sh file below. Notice the shebang (#!) line: it ensures the file can be executed by Bash.
Execute the queries
I executed the commands.sh file with the help of Python’s subprocess library. Notice I indicated the path of the file by converting the libpath.Path() address to a string with str() and changed directories with the cwd argument so that the queries results would be downloaded into the data folder as well.
subprocess.call(str(data_folder / "commands.sh"), cwd=data_folder)
Then I waited for the download completion. The print screen below shows part of the wget progress.
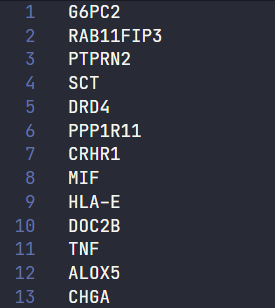
After a while, two files, named GO0002790.txt and GO0019932.txt were created, each containing a list of several genes related with the GO ids:
.
├── data
│ ├── commands.sh
│ ├── go_demo.xlsx
│ ├── GO0002790.txt
│ ├── GO0019932.txt
│ └── lists_ensembl.xlsx
├── src
│ ├── files_to_pandas.py
│ ├── lists_ensembl.R
│ └── query_biomart.py
└── ensembl_rest.py
Label and unify data into a pandas.DataFrame
Similarly to my previous post I needed to make a relation of GO id —> genes, so I adapted and renamed the old function I used on that occasion. The new function is named files_to_pandas. It takes a file and converts it into a pandas.DataFrame while creating a new column to accommodate the file name into a new column (in our case, the files were named after the values ‐ GO ids).
The idea is that I needed to create one data frame for each file. Thus, I used the glob method to get an iterable of file names and used dask‘s delayed method to assemble the several pandas.DataFrames generated by the files_to_pandas function loop into a unified dask.DataFrame.
file_list = data_folder.glob("*.txt")
dfs = [delayed(files_to_pandas)(filename) for filename in file_list]
ddf = dd.from_delayed(dfs)
Reorganize data with defaultdict
I could have ended in the previous step, but I surmised that I could condense the data frame into fewer rows, by aggregating all genes into the same row of the equivalent GO id. So I had the idea to use Python’s defaultdict. It is a subclass of the dictionary class. First, I initialize an empty defaultdict. It will have GO ids as keys and a list of gene names as values:
go_to_genes = defaultdict(list)
Then I simply had to loop through the rows of the dask.DataFrame, insert the keys into the defaultdict and appending the gene names into the value-list one by one.
for go_id, gene in zip(ddf["filename"], ddf["gene_name"]):
go_to_genes[go_id].append(gene)
Save the defaultdict into a new pandas.DataFrame
Finally, I created another data frame to store the contents of the defaultdict. Notice how I used the .items() method. It is the correct way to access the contents of a defaultdict.
go_genes = pd.DataFrame(go_to_genes.items(), columns=["go_ids", "gene_name"])
Error checking
Just to be sure that all queries worked correctly, a used a simple loop to check the contents of the gene names column:
for go_id, gene in zip(go_genes["go_ids"],go_genes["gene_name"]):
if "Query" in str(gene):
print(f"Error in {go_id}")
else:
continue
print("Error check done.")
I am using “Query” is used as an example, because BioMart errors may contain this word in their error messages, such as:
"Query ERROR: caught BioMart::Exception::Database: Could not connect to mysql database ..."
Of course, other strings can be used. In retrospect, “ERROR” would have been an even better option than “Query”. If some error were detected, the loop would print the GO id that failed the query (due to a network error, for example), so I would have to retry the query.
Annotate original data frame
No errors were found, so I finally could annotate my original data frame by joining them by the "go_ids" column. I even created a new column ("gene_string") to convert the Python lists into a nicely formatted comma-delimited string of gene names:
df_annotated = df.set_index("go_ids").join(go_genes.set_index("go_ids"))
df_annotated["gene_string"] = [", ".join(map(str, element)) for element in df_annotated["gene_name"]]
I then saved the result into a new tab (named go annotated) inside my go_demo.xlsx file:
with pd.ExcelWriter(go_list, mode="a") as writer:
df_annotated.to_excel(writer, sheet_name="go annotated", index=True)
Conclusion
In this post, I:
- Introduced a function to create customized ready-to-use query strings via REST API access;
- Provided a file with the descriptors of data deposited in BioMart (
list_ensembl.xlsx); - Demonstrated how to use Python to invoke
GNU Wgetto download the the queries’ results; - Demonstrated how to aggregate and manipulate the queries results using
pandas,daskanddefaultdict.
Feel free to use the query_biomart() function to data mine BioMart as you wish!
Subscribe to my RSS feed, Atom feed or Telegram channel to keep you updated whenever I post new content.
Appendix
I prepared a spreadsheet named lists_ensembl.xlsx also stored into the data folder. Ensembl has a lot of datasets, filters and attributes, so examine the other rows if you are interested. I produced the spreadsheet with the help of biomaRt R package. If you prefer R over Python, be sure to try querying BioMart with it. The lists_ensembl.R file contains the R code that generated the spreadsheet.
References
Wget - GNU Project - Free Software Foundation
Representational state transfer - Wikipedia
goana function | R Documentation
Working with Cancer Genomics Cloud datasets in a PostgreSQL database (Part 2)
pathlib — Object-oriented filesystem paths — Python 3.9.1 documentation
openpyxl - A Python library to read/write Excel 2010 xlsx/xlsm files — openpyxl 3.0.6 documentation
collections — Container datatypes | Python 3.9.1 documentation